Morgellons disease
Morgellons disease is an infectious skin condition that is linked to spirochetal (bacterial) infection and tick-borne illness disfiguring and disabling patients. The name came from an unrelated condition in France in 1674, in a letter written by English physician Sir Thomas Brown; he mentioned black hairs emerging on the backs of children in Languedoc. He called it Morgellons and said it cured their coughs and convulsions. A matching description was first reported in the US in 2002.
Morgellons patients suffer from feelings of itching, biting, and crawling sensations in the skin; microscopic filaments resembling textile fibers of colours black, white, blue or red growing from the skin; skin lesions, cognitive functions such as memory loss, poor concentration, joint pains, depressed moods, and fatigue.

The skin filaments have been identified as compressed cellular proteins such as keratin and collagen; an overproduction of these filaments is in response to a spirochetal infection. This infection affects humans, cattle, cats, and dogs. There were 14,000 reports in 2009; since then, there has been an increase in patients with Morgellons disease.
Research shows that skin afflictions were due to a psychological response to the presence of an infection. There is no known cure for Morgellons disease. Some doctors believe Morgellons to be a psychological condition and treat it with cognitive behavioral therapy, antidepressants, antipsychotics, and counseling. Other doctors believe it to be biological. Further studies are required to properly understand the condition.
Paraneoplastic pemphigus
Paraneoplastic Pemphigus (PNP) is a rare autoimmune skin disorder linked to the group of blistering skin diseases. It is a neoplasm. The first symptoms of PNP are usually oral, skin and mucosal lesions. The pathogenesis of pemphigus is still not completely understood, although some immunological characteristics have been recently reported.
Since it’s rare, several diagnostic criteria have been proposed and many diagnostic procedures have been used for diagnosis; they are indirect immunofluorescence and direct immunofluorescence, which are antibody tests using fluorescent dyes and ELISA, which is a test for detecting and quantifying antibodies and hormones.
PNP causes cancers in the stomach, lungs, and colon. B-cell Lymphoma and haematological malignancies are constantly associated with PNP. Patients can show multi-organ involvement and a subset of autoantibodies to several tissues.
Early diagnosis of PNP is difficult, but it’s the best option. Usually, a neoplasm is detected before the onset of PNP, however, PNP is the first clinical manifestation that leads to the discovery of underlying tumors and also that causes secretion of functional peptides and hormone which causes blisters on the skin and mucous membranes.

The blisters fill with fluid and peel off, leaving the skin raw and open and making it susceptible to infection. The disease is extremely fatal, causing death in 90% of patients diagnosed due to sepsis, cancer or multi-organ failure.
PNP usually affects people between ages of 45 and 70, but children and adolescents can be affected too. The disease is hard to treat; removal of tumors and treatment with steroids are the best options until a cure is possibly found. During the past few years, medical advances have improved the understanding of PNP pathogenesis and enhanced diagnosis. Effective diagnosis and treatment may affect the overall clinical outcome. Further research needs to be done.
Microcephaly
Microcephaly is defined by the circumference of the head being much smaller than usual, according to the gender and age of the child. There are two types of microcephaly: one is congenital microcephaly diagnosed at birth, while the other is postnatal microcephaly which appears later in life.
Genetic abnormalities, metabolic disorders, infections, teratogens, as well as prenatal, perinatal and postnatal injuries can cause congenital and postnatal microcephaly. Patients are evaluated with a thorough history and physical examination. Neuroimaging, metabolic, or genetic testing should be done in case of aggravation of microcephaly. Further diagnosis should be done owing to the underlying symptoms pointing to an underlying diagnosis.

Often the first tests for diagnosing microcephaly are Magnetic Resonance imaging (MRI) and neuroimaging. Genetic testing is becoming more common as a follow-up to neuroimaging when the physical examination has no successful diagnosis. Microcephaly is a lifelong illness with no known cure; the prognosis is usually worse for children with intrauterine infection, or metabolic or chromosomal abnormality.
Maternal infections and the Zika virus are associated with microcephaly and other serious brain abnormalities. Children affected by microcephaly end up with less or no brain development while in the womb due to genetic abnormalities and interference in the growth of the cerebral cortex. This causes the child to develop a smaller-than-usual head at birth, which usually remain the same during growth.
Many believe the reasons to be genetic or exposure to harmful substances like radiation, drugs, alcohol, or certain viruses like the Zika virus, rubella, (German measles), varicella (chickenpox), the cytomegalic virus while in the womb, or untreated phenylketonuria (PKU).
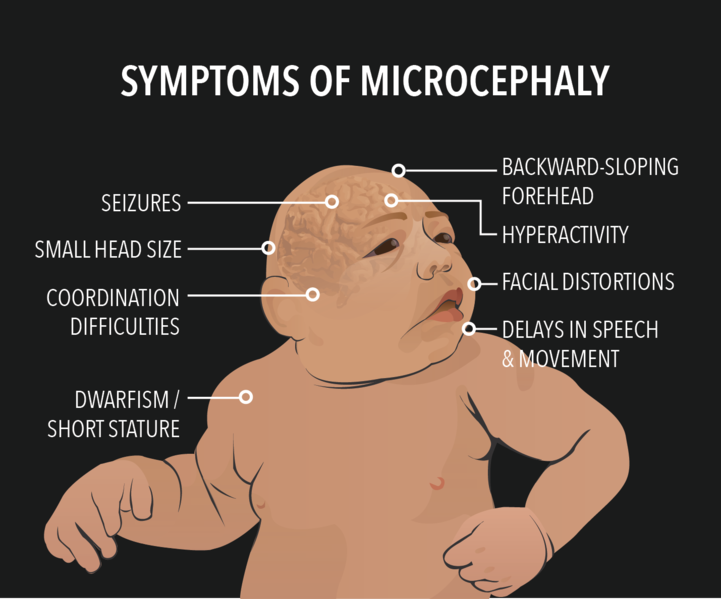
The disease is usually paired with Down syndrome. A person affected by Microcephaly ends up with issues of intellectual disability, delayed development, cerebral palsy, seizures, as well as hearing and visual issues. Rubella and Varicella are other serious conditions known to cause genetic microcephaly and intellectual deficits. Maternal immunization for rubella and varicella and have known to prevent microcephaly.
There is no cure to get the child’s head to return to the correct size or shape, but treatment focuses on decreasing the impact associated with the deformities and neurological difficulties. Medicine is used to control seizures, hyperactivity, and neuromuscular symptoms. Genetic testing may help families in future pregnancies.
Von Hippel-Lindau Syndrome (VHL)
Von Hippel-Lindau Syndrome (VHL) is an extremely rare genetic condition which more frequently appears in young adulthood, but symptoms may persist throughout life as well. VHL causes cancerous or non-cancerous tumors or cysts (fluid-filled sacs) to grow in different parts of the body. These tumors are medically known as haemangioblastomas. They are made of newly-formed blood vessels which are more often benign.
These tumors grow in the retina of the eye (retinal angiomas), causing vision loss; as well as the brain and spinal cord, causing weakness, headaches, vomiting, and muscle coordination (ataxia). Cysts tend to grow in the pancreas, kidneys, and genital tract. They are at increased risk of developing kidney cancer called Clear Cell Renal Cell Carcinoma as well as pancreatic cancer called a Pancreatic Neuroendocrine Tumor.

VHL is also associated with a tumor known as pheochromocytoma, which is most commonly associated with adrenal glands (small glands located on top of each kidney which produces hormones). Pheochromocytomas are mostly benign. These tumors rarely show symptoms, but in some cases, they cause panic attacks, excessive sweating, headaches, and dangerously high blood pressure, which may not respond to medication.
Pheochromocytomas can be really dangerous in times of stress and trauma, like when associated with pregnancy, accidents, or surgery. Around 10% of people with HPL develop endolymphatic sac tumors; non-cancerous inner ear tumors. These tumors can cause hearing loss in one or both ears, causing ringing in the ears (Tinnitus) and balance issues. Left untreated, these tumors can cause sudden and profound deafness.
There is a chance of non-cancerous tumors developing in the lungs or liver with people who have VHL, but they may not show any symptoms.
Fibrodysplasia Ossificans Progressiva (FOP)
FOP is a progressive disorder in which muscle tissue and connective tissue such as tendons and ligaments are replaced by bones (ossified), forming bones outside the skeleton (extraskeletal or heterotopic bones) that affect bone movement.
FOP affects the joints, causing loss of mobility, as well as inability to open the mouth (making it impossible to eat or speak, which leads to malnutrition). Extra bone formation around the rib cage can also cause breathing difficulties.
This condition is so rare that it said to affect one in two million people worldwide. There have been several hundred cases reported. FOP is first noticed at early childhood, which starts at the neck and shoulders and then proceeds down to the body and limbs.

Injury or invasive muscle procedures can aggravate the process, causing episodes of inflammation and muscle swelling, which speeds up ossification in the injured area. Flare-ups can also occur due to viral infections such as influenza.
At birth, there is a classic symptom known as malformation of the big toes. This condition can cause short thumbs and other skeletal abnormalities too. There is no known treatment for FOP as surgery to remove the bone can cause the body to produce even more.
No comments:
Post a Comment